| <<< Previous | Index | Next >>> |
Case 7
Clinical and haematological data
67-yr-old man. Presents now with marked splenomegaly and anemia.
A diagnosis of Polycythemia Vera was made 12 years ago.
No hepatomegaly, no adenopathy. Hb 100g/l, L 10 G/l, T 160 G/l
On PB smears: erythroblasts 3%, myelocyte 0.5%. RBC aniso-poikilocytosis with tear-drop shaped RBCs.
BM aspirate: dry tap.
BM biopsy. Histological examination disclose an hypercellular marrow with strong hyperplasia of the megakaryocytic cell line. The megakaryocytes form large clusters and present variable morphological features. Most of them exhibit large and hyperlobulated nuclei but some small forms can also been observed characterized by dark and hyperchromatic nuclei and poor cytoplasm. The erythroid cell line is present with rare dysplastic changes. The granulocytic cell line predominates with good maturation and no shift to the left. No excess of blast cells. Diffuse increase in reticulin (Bauermeister grade 2). The marrow sinusoid are distended and contain foci of haemopoietic cells. No excess of CD34+ cells at
IHC .
Interpretation and diagnosis (taking in account the clinical history and the haematological data) : Postpolycythemia Myeloid Metaplasia (PPMM) or Myelofibrosis with myeloid metaplasia supervening during the course of PV.
Comments
The BM picture is virtually indistinguishable from that of Chronic Idiopathic or Primary Myelofibrosis in cellular phase. This case underlines the importance of careful correlation of clinical, laboratory, and histological findings in reaching the best diagnosis.
Approximately 15 % of patients with PV develop Myelofibrosis or PPMM also termed "spent phase of PV". This phase appears after an average interval of 10 years from diagnosis. The incidence among patients with PV who survive for 15 years or more is reported to approach 50%. For some time it has been thought that the use of radioactive phosphorus (
32 P) in the treatment of PV was associated with an increase incidence of PPMM. However, this was not confirmed by the results of the PVSG (Polycythemia Vera Study Group). Patients with PPMM are characterized by splenomegaly, leukoerythroblastosis, teardrop RBC poikilocytosis, normalization or decrease of the RBC mass, BM failure and extensive marrow fibrosis. The onset of PPMM is associated with a shorter survival than for patients with primary or idiopathic myelofibrosis. Most patients die in less than 3 years from the diagnosis of PPMM and transformation to acute leukaemia is a frequent cause of death. Approximately 1% to 2% of PV patients treated with phlebotomy alone develop acute leukaemia. However, the frequency is higher (10% to 15%) in patients treated with myelosuppressive agents and in nearly 50% of these patients, the blastic transformation is preceded by a myelodysplastic period. Thus, the finding of myelodysplastic features in the follow-up biopsies of PV patients should be viewed with concern.
The differential diagnosis of myelofibrosis includes: leukoerythroblastosis due to a metastatic process involving BM, myelodysplastic syndromes (MDS) with fibrosis (possibly therapy-related MDS). According to the literature, patients with MDS and fibrosis are characterized by minimal organomegaly and prominent dysplasia in megakaryopoiesis. They also have a higher frequency of cytogenetic aberrations.
The case presented here illustrates the diagnostic and prognostic value of BM biopsy in the assessment of MPD at presentation and during follow-up.
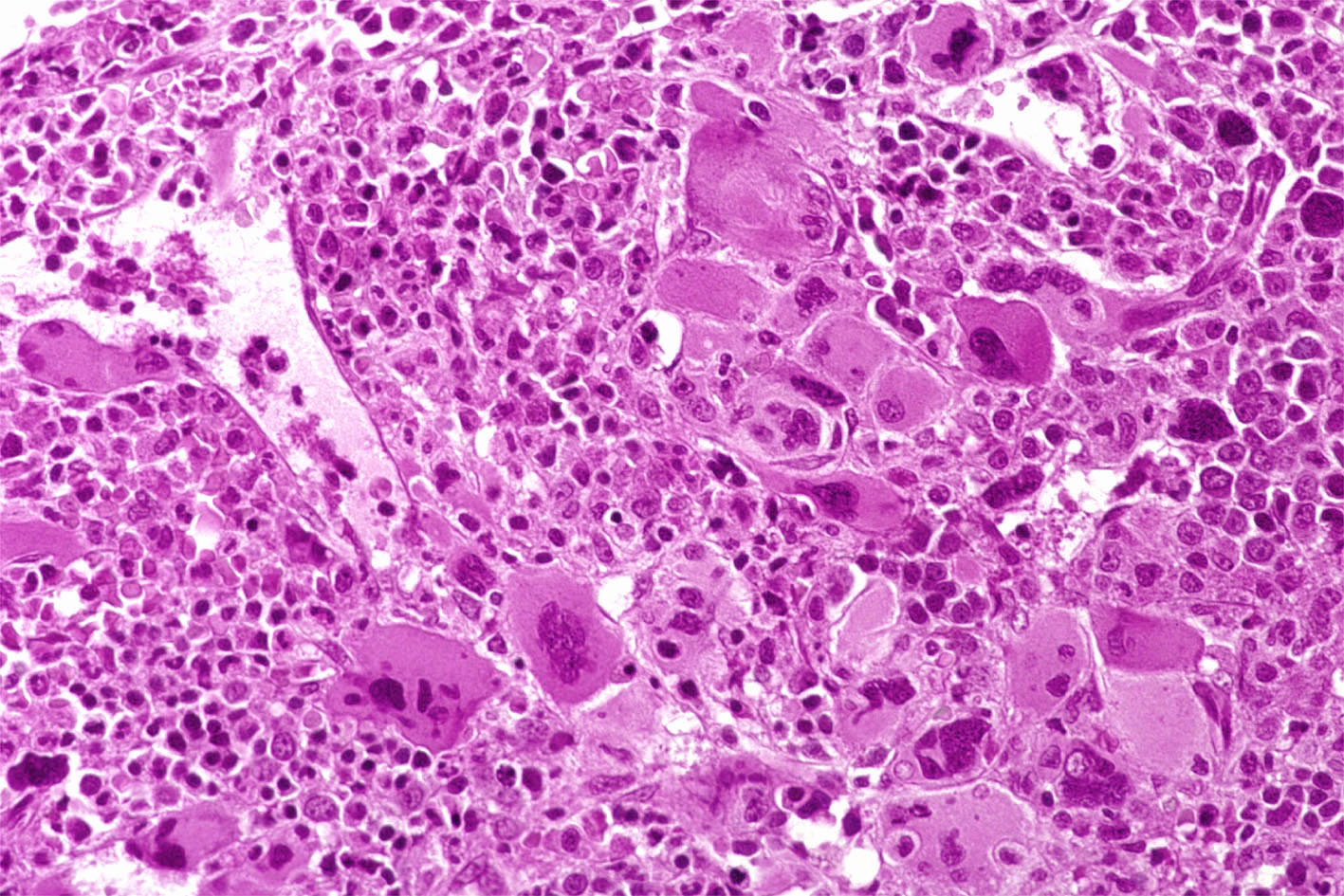
Case 7. Postpolycythemia Myeloid Metaplasia.
| <<< Previous | Index | Next >>> |
Copyright 2001, The Author(s) and/or The Publisher(s)
| Organisation: FORPATH asbl |
Coordination: Dr Bernard Van den Heule |
Host: Labo CMP |